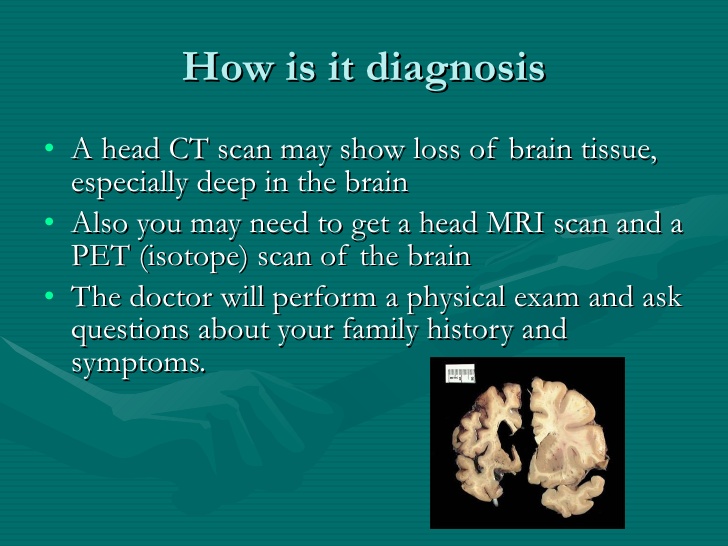
The National Institute of aging states the following procedures also may be used to diagnose dementia:
“Cognitive and neuropsychological tests. These tests are used to assess memory, problem solving, language skills, math skills, and other abilities related to mental functioning.
Laboratory tests. Testing a person’s blood and other fluids , as well as checking levels of various chemicals, hormones, and vitamins, can help find or rule out possible causes of symptoms.
Brain scans. These tests can identify strokes, tumors, and other problems that can cause dementia. Scans also identify changes in the brain’s structure and function. The most common scans are:
Computed tomography (CT), which uses x rays to produce images of the brain and other organs
Magnetic resonance imaging (MRI), which uses magnetic fields and radio waves to produce detailed images of body structures, including tissues, organs, bones, and nerves
Positron emission tomography (PET), which uses radiation to provide pictures of brain activity
Psychiatric evaluation. This evaluation will help determine if depression or another mental health condition is causing or contributing to a person’s symptoms.
Genetic tests. Some dementias are caused by a known gene defect. In these cases, a genetic test can help people know if they are at risk for dementia. It is important to talk with a genetic counselor before and after getting tested, along with family members and the doctor.
“Alzheimer’s disease is a type of brain disease, just as coronary artery disease is a type of heart disease. It is
caused by damage to nerve cells (neurons) in the brain. The brain’s neurons are essential to thinking, walking,
talking and all human activity. In Alzheimer’s, the neurons damaged first are those in parts of the brain responsible for memory, language and thinking. As a result, the first symptoms tend to be memory,
language and thinking problems. Although these symptoms are new to the individual affected, the brain changes that cause them are thought to begin 20 years or more before symptoms start.”.
Alzheimer’s Association (https://www.alz.org › alzheimers-facts-and-figures)
What structurally happens to the brain and what happens to the individual in brain thinking diagnosed with this disease:
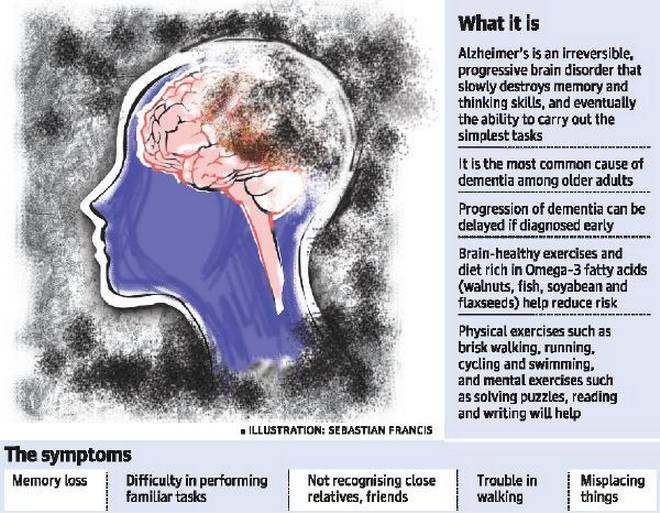
Alzheimer’s disease is an irreversible, progressive brain disorder that slowly destroys memory and thinking skills and, eventually, the ability to carry out the simplest tasks. In most people with the disease—those with the late-onset type—symptoms first appear in their mid-60s. Early-onset Alzheimer’s occurs between a person’s 30s and mid-60s and is very rare. Alzheimer’s disease is the most common cause of dementia among older adults.
The disease is named after Dr. Alois Alzheimer. In 1906, Dr. Alzheimer noticed changes in the brain tissue of a woman who had died of an unusual mental illness. Her symptoms included memory loss, language problems, and unpredictable behavior. After she died, he examined her brain and found many abnormal clumps (now called amyloid plaques) and tangled bundles of fibers (now called neurofibrillary, or tau, tangles).
These plaques and tangles in the brain are still considered some of the main features of Alzheimer’s disease. Another feature is the loss of connections between nerve cells (neurons) in the brain. Neurons transmit messages between different parts of the brain, and from the brain to muscles and organs in the body. Many other complex brain changes are thought to play a role in Alzheimer’s, too.
This damage initially appears to take place in the hippocampus, the part of the brain essential in forming memories. As neurons die, additional parts of the brain are affected. By the final stage of Alzheimer’s, damage is widespread, and brain tissue has shrunk significantly.
How many affected in the United States by Alzheimer’s Disease:
Estimates vary, but experts suggest that as many as 5.5 million Americans age 65 and older may have Alzheimer’s. Many more under age 65 also have the disease. Unless Alzheimer’s can be effectively treated or prevented, the number of people with it will increase significantly if current population trends continue. This is because increasing age is the most important known risk factor for Alzheimer’s disease.
Symptoms:
Memory problems are typically one of the first signs of Alzheimer’s, though initial symptoms may vary from person to person. A decline in other aspects of thinking, such as finding the right words, vision/spatial issues, and impaired reasoning or judgment, may also signal the very early stages of Alzheimer’s disease. Mild cognitive impairment (MCI) is a condition that can be an early sign of Alzheimer’s, but not everyone with MCI will develop the disease.
People with Alzheimer’s have trouble doing everyday things like driving a car, cooking a meal, or paying bills. They may ask the same questions over and over, get lost easily, lose things or put them in odd places, and find even simple things confusing. As the disease progresses, some people become worried, angry, or violent.
Alzheimer’s disease is not a normal part of aging.
Memory problems are typically one of the first warning signs of cognitive loss.
According to the National Institute on Aging, in addition to memory problems, someone with Alzheimer’s disease may experience one or more of the following signs:
Memory loss that disrupts daily life, such as getting lost in a familiar place or repeating questions.
Trouble handling money and paying bills.
Difficulty completing familiar tasks at home, at work or at leisure.
Decreased or poor judgment.
Misplaces things and being unable to retrace steps to find them.
“The Lupus Foundation of America estimates that 1.5 million Americans, and at least five million people worldwide, have a form of lupus! Lupus strikes mostly women of childbearing age. Ninety percent (90%) of people living with lupus are women. However, men, children, and teenagers develop lupus, too. Most people with lupus develop the disease between the ages of 15-44. Most people with lupus develop the disease between the ages of 15-44. Lupus is two to three times more prevalent among African American, Hispanic/Latina, Asian American, Native American, Alaska Native, Native Hawaiian and other Pacific Islander women than among White women.”.
Lupus Foundation of America (https://www.lupus.org/resources/lupus-facts-and-statistics)
“World Lupus Day was 5/10/23 and by WELCOA a health topic for May. Genes do play a role in the predisposition to the development of lupus. There are dozens of known genetic variants linked to lupus. These genes impact both who gets lupus and how severe it is. 20 percent of people with lupus will have a parent or sibling who already has lupus or may develop lupus. About 5 percent of the children born to individuals with lupus will develop the illness. Although lupus can develop in people with no family history of lupus, there are likely to be other autoimmune diseases in some family members. Lupus is not contagious, not even through sexual contact. You cannot “catch” lupus from someone or “give” lupus to someone.
World Lupus Day (https://worldlupusday.org/lupus-facts-and-statistics/)
“You can’t cure or slow the progression of Huntington disease, but health care providers can offer medications to help with certain symptoms. As Huntington disease progresses, you will need constant assistance and supervision because of the debilitating nature of the disease. People usually die from the disease within 15 to 20 years of developing symptoms. If you have been diagnosed with, or are at risk for Huntington disease, it is critical to maintain your physical fitness as best you can. People who exercise regularly and stay active tend to do better than those who don’t. A number of studies are currently under way to examine possible therapies for Huntington disease.”
John Hopkins Hospital (https://www.hopkinsmedicine.org/health/conditions-and-diseases/huntingtons-disease)
No treatments can alter the course of Huntington’s disease. But medications can lessen some symptoms of movement and psychiatric disorders. And multiple interventions can help a person adapt to changes in his or her abilities for a certain amount of time.
Medications will likely evolve over the course of the disease, depending on overall treatment goals. Also, drugs that treat some symptoms may result in side effects that worsen other symptoms. Treatment goals will be regularly reviewed and updated.
Preparing for your doctor’s appointment
If you have any signs or symptoms associated with Huntington’s disease, you’ll likely be referred to a neurologist after an initial visit to your family doctor.
A review of your symptoms, mental state, medical history and family medical history can all be important in the clinical assessment of a potential neurological disorder.
What you can do
Before your appointment, make a list that includes the following:
Signs or symptoms — or any changes from what is normal for you — that may be causing concern
Recent changes or stresses in your life
All medications — including over-the-counter drugs and dietary supplements — and doses you take
Family history of Huntington’s disease or other disorders that may cause movement disorders or psychiatric conditions
You may want a family member or friend to accompany you to your appointment. This person can provide support and offer a different perspective on the effect of symptoms on your functional abilities.
What to expect from your doctor
Your doctor is likely to ask you a number of questions, including the following:
When did you begin experiencing symptoms?
Have your symptoms been continuous or intermittent?
Has anyone in your family ever been diagnosed with Huntington’s disease?
Has anyone in your family been diagnosed with another movement disorder or psychiatric disorder?
Are you having trouble performing work, schoolwork or daily tasks?
Has anyone in your family died young?
Is anyone in your family in a nursing home?
Is anyone in your family fidgety or moving all the time?
Have you noticed a change in your general mood?
Do you feel sad all of the time?
Have you ever thought about suicide?
Medications used for Huntington’s Disease
Medications for movement disorders
Drugs to treat movement disorders include the following:
Drugs to control movement include tetrabenazine (Xenazine) and deutetrabenazine (Austedo), which have been specifically approved by the Food and Drug Administration to suppress the involuntary jerking and writhing movements (chorea) associated with Huntington’s disease. These drugs don’t have any effect on the progression of the disease, however. Possible side effects include drowsiness, restlessness, and the risk of worsening or triggering depression or other psychiatric conditions.
Antipsychotic drugs, such as haloperidol (Haldol) and fluphenazine, have a side effect of suppressing movements. Therefore, they may be beneficial in treating chorea. However, these drugs may worsen involuntary contractions (dystonia), restlessness and drowsiness.Other drugs, such as risperidone (Risperdal), olanzapine (Zyprexa) and quetiapine (Seroquel), may have fewer side effects but still should be used with caution, as they may also worsen symptoms.
Other medications that may help suppress chorea include amantadine (Gocovri ER, Osmolex ER), levetiracetam (Keppra, Elepsia XR, Spritam) and clonazepam (Klonopin). However, side effects may limit their use.
Medications for psychiatric disorders
Medications to treat psychiatric disorders will vary depending on the disorders and symptoms. Possible treatments include the following:
Antidepressants include such drugs as citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac, Sarafem) and sertraline (Zoloft). These drugs may also have some effect on treating obsessive-compulsive disorder. Side effects may include nausea, diarrhea, drowsiness and low blood pressure.
Antipsychotic drugs such as quetiapine (Seroquel), risperidone (Risperdal) and olanzapine (Zyprexa) may suppress violent outbursts, agitation, and other symptoms of mood disorders or psychosis. However, these drugs may cause different movement disorders themselves.
Mood-stabilizing drugs that can help prevent the highs and lows associated with bipolar disorder include anticonvulsants, such as divalproex (Depakote), carbamazepine (Carbatrol, Epitol, others) and lamotrigine (Lamictal).
Types of therapies for Huntington’s Disease
Psychotherapy
A psychotherapist — a psychiatrist, psychologist or clinical social worker — can provide talk therapy to help with behavioral problems, develop coping strategies, manage expectations during progression of the disease and facilitate effective communication among family members.
Speech therapy
Huntington’s disease can significantly impair control of muscles of the mouth and throat that are essential for speech, eating and swallowing. A speech therapist can help improve your ability to speak clearly or teach you to use communication devices — such as a board covered with pictures of everyday items and activities. Speech therapists can also address difficulties with muscles used in eating and swallowing.
Physical therapy
A physical therapist can teach you appropriate and safe exercises that enhance strength, flexibility, balance and coordination. These exercises can help maintain mobility as long as possible and may reduce the risk of falls.
Instruction on appropriate posture and the use of supports to improve posture may help lessen the severity of some movement problems.
When the use of a walker or wheelchair is required, the physical therapist can provide instruction on appropriate use of the device and posture. Also, exercise regimens can be adapted to suit the new level of mobility.
Occupational therapy
An occupational therapist can assist the person with Huntington’s disease, family members and caregivers on the use of assistive devices that improve functional abilities. These strategies may include:
Handrails at home
Assistive devices for activities such as bathing and dressing
Eating and drinking utensils adapted for people with limited fine motor skills
Lifestyle and home remedies
Managing Huntington’s disease is demanding on the person with the disorder, family members and other in-home caregivers. As the disease progresses, the person will become more dependent on caregivers. A number of issues will need to be addressed, and strategies to cope with them will evolve.
Eating and nutrition
Factors regarding eating and nutrition include the following:
Difficulty maintaining a healthy body weight. Difficulty eating, higher caloric needs due to physical exertion or unknown metabolic problems may be the cause. To get adequate nutrition, you may need to eat more than three meals a day or use dietary supplements.
Difficulty with chewing, swallowing and fine motor skills. These problems can limit the amount of food you eat and increase the risk of choking. Problems may be minimized by removing distractions during a meal and selecting foods that are easier to eat. Utensils designed for people with limited fine motor skills and covered cups with straws or drinking spouts also can help.
Eventually, a person with Huntington’s disease will need assistance with eating and drinking.
Managing cognitive and psychiatric disorders
Family and caregivers can help create an environment that may help a person with Huntington’s disease avoid stressors and manage cognitive and behavioral challenges. These strategies include:
Using calendars and schedules to help keep a regular routine
Initiating tasks with reminders or assistance
Prioritizing or organizing work or activities
Breaking down tasks into manageable steps
Creating an environment that is as calm, simple and structured as possible
Identifying and avoiding stressors that can trigger outbursts, irritability, depression or other problems
For school-age children or adolescents, consulting with school staff to develop an appropriate individual education plan
Providing opportunities for the person to maintain social interactions and friendships as much as possible
Coping and support
A number of strategies may help people with Huntington’s disease and their families cope with the challenges of the disease.
Support services
Support services for people with Huntington’s disease and families include the following:
Nonprofit agencies, such as the Huntington’s Disease Society of America, provide caregiver education, referrals to outside services, and support groups for people with the disease and caregivers.
Local and state health or social service agencies may provide daytime care for people with the disease, meal assistance programs or respite for caregivers.
Planning for residential and end-of-life care
Because Huntington’s disease causes the progressive loss of function and death, it’s important to anticipate care that will be needed in the advanced stages of the disease and near the end of life. Early discussions about this type of care enable the person with Huntington’s disease to be engaged in these decisions and to communicate his or her preferences for care.
Creating legal documents that define end-of-life care can be beneficial to everyone. They empower the person with the disease, and they may help family members avoid conflict late in the disease progression. Your doctor can offer advice on the benefits and drawbacks of care options at a time when all choices can be carefully considered.
Matters that may need to be addressed include:
Care facilities. Care in the advanced stages of the disease will likely require in-home nursing care or care in an assisted living facility or nursing home.
Hospice care. Hospice services provide care at the end of life that helps a person approach death with as little discomfort as possible. This care also provides support and education to family members to help them understand the process of dying.
Living wills. Living wills are legal documents that enable a person to spell out care preferences when he or she isn’t able to make decisions. For example, these directions might indicate whether or not the person wants life-sustaining interventions or aggressive treatment of an infection.
Advance directives. These legal documents enable you to identify one or more people to make decisions on your behalf. You may create an advance directive for medical decisions or financial matters.
“Huntington Disease (HD) typically begins between the ages of 30-45, though onset may occur as early as the age of two or as late as the 70s. HD affects males and females equally and affects all ethnic and racial groups. Symptoms of HD can differ from person to person, even for members of the same family. Early symptoms may include depression, mood swings, forgetfulness, clumsiness, involuntary twitching, and lack of coordination. HD usually progresses over a 10-25 year period. Death follows from complications such as choking or infection. Affected individuals require assistance for daily care such as bathing and dressing. Each child of a person with HD has a 50% chance of inheriting the HD-causing gene mutation. More than 250,000 Americans have HD or are “at-risk” of inheriting the disease from an affected parent. A blood test can accurately determine whether an adult carries the HD-causing gene.”
A preliminary diagnosis of Huntington’s disease is based primarily on your answers to questions, a general physical exam, a review of your family medical history, and neurological and psychiatric examinations.
Neurological examination
The neurologist will ask you questions and conduct relatively simple tests of your:
Motor symptoms, such as reflexes, muscle strength and balance
Sensory symptoms, including sense of touch, vision and hearing
Psychiatric symptoms, such as mood and mental status
Neuropsychological testing
The neurologist may also perform standardized tests to check your:
Memory
Reasoning
Mental agility
Language skills
Spatial reasoning
Psychiatric evaluation
You’ll likely be referred to a psychiatrist for an examination to look for a number of factors that could contribute to your diagnosis, including:
Emotional state
Patterns of behaviors
Quality of judgment
Coping skills
Signs of disordered thinking
Evidence of substance abuse
Brain imaging and function
Your doctor may order brain-imaging tests for assessing the structure or function of the brain. The imaging technologies may include MRI or CT scans that show detailed images of the brain.
These images may reveal changes in the brain in areas affected by Huntington’s disease. These changes may not show up early in the course of the disease. These tests can also be used to rule out other conditions that may be causing symptoms.
Genetic counseling and testing
If symptoms strongly suggest Huntington’s disease, your doctor may recommend a genetic test for the defective gene.
This test can confirm the diagnosis. It may also be valuable if there’s no known family history of Huntington’s disease or if no other family member’s diagnosis was confirmed with a genetic test. But the test won’t provide information that might help determinine a treatment plan.
Before having such a test, the genetic counselor will explain the benefits and drawbacks of learning test results. The genetic counselor can also answer questions about the inheritance patterns of Huntington’s disease.
Predictive genetic test
A genetic test can be given if you have a family history of the disease but don’t have symptoms. This is called predictive testing. The test can’t tell you when the disease will begin or what symptoms will appear first.
Some people may have the test because they find not knowing to be more stressful. Others may want to take the test before having children.
“Know Huntington’s disease is a rare, inherited disease that causes the progressive breakdown (degeneration) of nerve cells in the brain. Huntington’s disease has a wide impact on a person’s functional abilities and usually results in movement, thinking (cognitive) and psychiatric disorders. Huntington’s disease symptoms can develop at any time, but they often first appear when people are in their 30s or 40s. If the condition develops before age 20, it’s called juvenile Huntington’s disease. When Huntington’s develops early, symptoms are somewhat different and the disease may progress faster. Medications are available to help manage the symptoms of Huntington’s disease. But treatments can’t prevent the physical, mental and behavioral decline associated with the condition.”