Should focus attention on important health messages about gastroparesis diagnosis, treatment, and quality of life issues. The goals include improving understanding of gastroparesis to help patients and families manage the condition, and encouraging preventive strategies.
The number of people with gastroparesis appears to be rising. Yet gastroparesis is poorly understood. More community awareness is needed about the condition.
The more awareness for gastroparesis, the greater the ability to impact positive outcomes, such as additional research and improved patient care for the functional GI and motility disorders.
Gastroparesis is also called delayed gastric emptying. The term “gastric” refers to the stomach.
Normally, the stomach empties its contents in a controlled manner into the small intestines. In gastroparesis, the muscle contractions (motility) that move food along the digestive tract do not work properly and the stomach empties too slowly.
Gastroparesis is characterized by the presence of certain long-term symptoms together with delayed stomach emptying in the absence of any observable obstruction or blockage. The delayed stomach emptying is confirmed by a test.
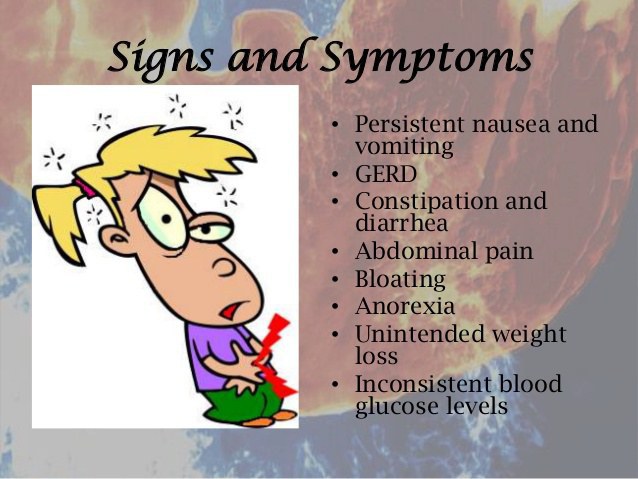
Signs and Symptoms:
The signs and symptoms of gastroparesis may differ among persons with the condition. Symptoms usually occur during and after eating a meal.
Symptoms that are characteristic of gastroparesis include:
- Nausea and/or vomiting
- Retching (dry heaves)
- Stomach fullness after a normal sized meal
- Early fullness (satiety) – the inability to finish a meal
Diagnosing Gastroparesis:
The symptoms of gastroparesis are similar to those that occur in a number of other illnesses. When symptoms persist over time or keep coming back, it’s time to see a doctor to diagnose the problem. An accurate diagnosis is the starting point for effective treatment.
Diagnosis of gastroparesis begins with a doctor asking about symptoms and past medical and health experiences (history), and then performing a physical exam. Any medications that are being taken need to be disclosed.
Tests will likely be performed as part of the examination. These help to identify or rule out other conditions that might be causing symptoms. Tests also check for anything that may be blocking or obstructing stomach emptying. Examples of these tests include:
- a blood test,
- an upper endoscopy, which uses a flexible scope to look into the stomach,
- an upper GI series that looks at the stomach on an x-ray, or
- an ultrasound, which uses sound waves that create images to look for disease in the pancreas or gallbladder that may be causing symptoms.
If – after review of the symptoms, history, and examination – the doctor suspects gastroparesis, a test to measure how fast the stomach empties is required to confirm the diagnosis.
Slow gastric emptying alone does not correlate directly with a diagnosis of gastroparesis.
There are several different ways to measure the time it takes for food to empty from the stomach into the small intestine. These include scintigraphy, wireless motility capsule, or breath test. Your doctor will provide details of the one chosen.
Gastric Emptying Study (Scintigraphy)
The diagnostic test of choice for gastroparesis is a gastric emptying study (scintigraphy). The test is done in a hospital or specialty center.
It involves eating a bland meal of solid food that contains a small amount of radioative material so that it can be tracked inside the body. The abdomen is scanned over the next few hours to see how quickly the meal passes out of the stomach. A radiologist will interpret the study at periodic intervals after the meal.
A diagnosis of gastroparesis is confirmed when 10% or more of the meal is still in the stomach after 4 hours.
Other methods for measuring gastric emptying include a wireless motility capsule and a breath test.
Wireless Motility Capsule
The ingestible wireless motility capsule (SmartPill) is swallowed and transmits data to a small receiver that the patient carries. The data collected is interpreted by a radiologist. While taking the test, people can go about their daily routine. After a day or two, the disposable capsule is excreted naturally from the body.
Breath Test
The breath test involves eating a meal that contains a nonradioactive component that can be tracked and measured in the breath over a period of hours. The results can then be calculated to determine how quickly the stomach empties.
Stay tune for Part II tomorrow!