

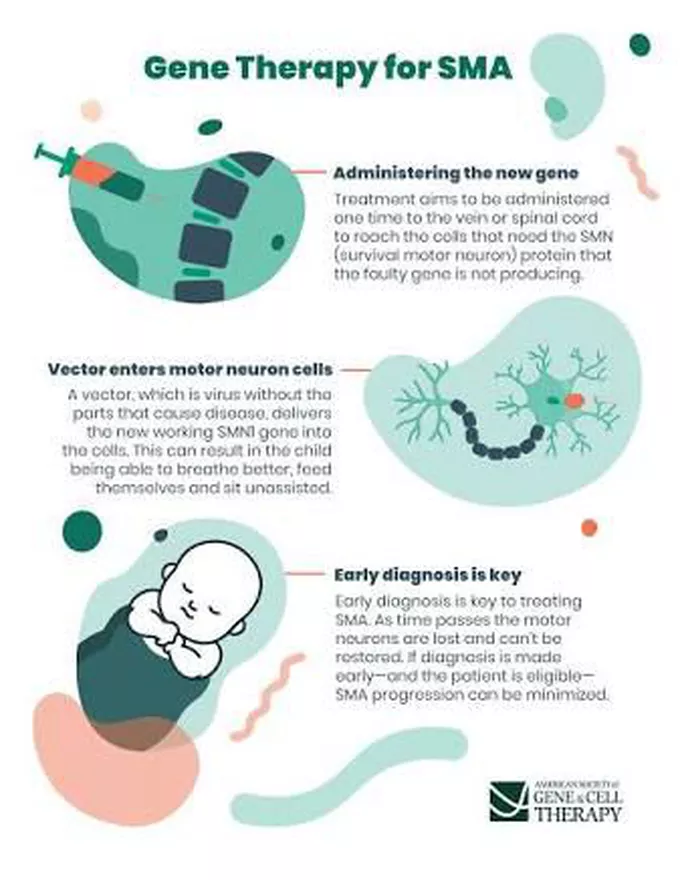
Research Trials for SMA leading to actual care for SMA:
The research picture has brightened considerably in the last decade for people with chromosome 5-related spinal muscular atrophy (SMA) types 0 through 4.
Since 1995, scientists have known that a deficiency of functional SMN protein (SMN stands for survival of motor neuron) is the underlying cause of chromosome 5 SMA. Two nearly identical genes carry the genetic instructions for making SMN protein: SMN1 and SMN2. Proteins made from the SMN1 gene are full-length, functional, and appear to be necessary for the survival and proper function of motor neurons. By contrast, proteins made using instructions from the SMN2 gene are shorter and tend to be less stable but can compensate for a lack of SMN protein when the SMN1 gene is not functioning.
In SMA types 0 through 4, flaws (mutations) in each of the two copies of the SMN1 genes result in insufficient production of full-length, functional SMN protein. Fortunately, a certain amount of full-length SMN protein can be made from the SMN2 gene. Many people have multiple copies of the SMN2 gene. These extra SMN2 copies can lessen the impact of a flaw in both SMN1 copies. In chromosome 5-related SMA, the more copies of SMN2 a person has, the milder the course of SMA is likely to be.
Researchers are seeking to exploit this unique redundancy through development of strategies that restore levels of full-length SMN protein.
1- Nusinersen is a drug that has been shown to increase the survival of motor neurons that die off in SMA, robbing children of muscle control. The drug compensates for the effects of the SMN1 mutation by rallying a “backup” gene, known as SMN2. SMN2, like SMN1, also makes the SMN protein needed to keep motor neurons healthy, but most of it is truncated and nonfunctional. Nusinersen uses a genetically based technology called antisense oligonucleotide to shore up this backup gene. This enables people to make more of the full-length, functional SMN protein.
2-Risdiplam is an oral drug taken daily that also appears to boost the production of the SMN protein through the SMN2 gene. Our researchers are an essential part of the clinical testing of risdiplam in the FIREFISH and JEWELFISH studies, which both continue to look at the drug’s efficacy and side effects in infants through adults.
The FIREFISH study involves infants age 1 month to 7 months with infantile-onset SMA, the most severe type. The babies receive risdiplam given as an oral drug daily. The drug increases SMN protein in the infants and improves their ability to sit without support. After two years of treatment, the FIREFISH study shows that 59 percent of treated babies were able to sit without support for five seconds, 65 percent maintained upright head control, 29 percent were able to turn over, and 30 percent were able to stand with support or supporting weight.
One-year results from the JEWELFISH study of risdiplam in children with all types of SMA aged 6 months to 60 years and previously treated with other SMA therapies, showed that risdiplam increases SMN protein levels. The drug appears to double SMN levels among patients who were previously treated with nusinerson or Zolgensma, highlighting its potential as an alternative or add-on therapy to those drugs.
3-Gene Therapy Trials: To learn more about the potential for gene replacement in children with SMA, we have been involved in several clinical studies. The first, the STR1VE trial, studied intravenous administration of the therapy in infants less than 7 months of age with type 1 SMA. This trial, which followed infants until they were 18 months old, provided the data used by the FDA to approve Zolgensma for the treatment of babies under 2 years.
The STRONG study involves intrathecal (inside the spinal canal) administration of the gene therapy for children between the ages of 6 months to 5 years with SMA type 2. STRONG is on an FDA-hold pending evaluation of pre-clinical information.
Treatment for SMA:
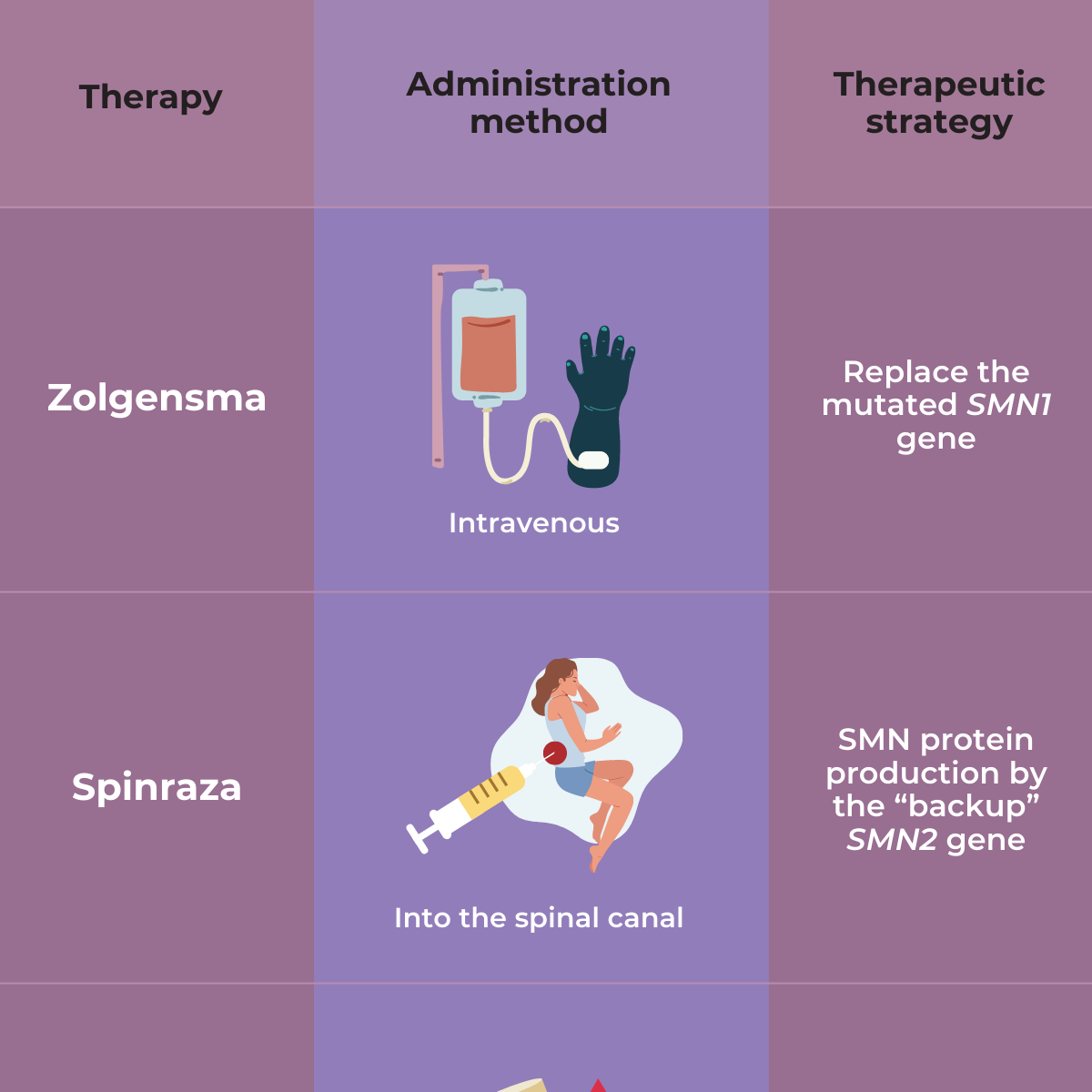
As yet, there is no complete cure for SMA. However, the discovery of the genetic cause of SMA has led to the development of several treatment options that affect the genes involved in SMA — a gene replacement therapy called Zolgensma, and two drugs, called nusinersen (Spinraza) and risdiplam (Evyrsdi).
There is no complete cure for SMA. Treatment consists of managing the symptoms and preventing complications.
Medications
- The U.S. Food and Drug Administration (FDA) approved nusinersen (Spinraza™) as the first drug approved to treat children and adults with SMA. The drug is designed to increase production of the SMN protein, which is critical for the maintenance of motor neurons.
- The FDA approved onasemnogene abeparovec-xioi (Zolgensma ™) gene therapy for children less than two years old who have infantile-onset SMA. A safe virus delivers a fully functional human SMN gene to the targeted motor neurons, which in turn improves muscle movement and function and survival.
- The FDA approved the orally-administered drug risdiplam (Evrysdi) to treat patients age two months of age and older with SMA.
Physical therapy, occupational therapy, and rehabilitation may help to improve posture, prevent joint immobility, and slow muscle weakness and atrophy. Stretching and strengthening exercises may help reduce contractures, increase range of motion, and keeps circulation flowing. Some individuals require additional therapy for speech and swallowing difficulties. Assistive devices such as supports or braces, orthotics, speech synthesizers, and wheelchairs may be helpful to improve functional independence.
Proper nutrition and calories are essential to maintaining weight and strength, while avoiding prolonged fasting. People who cannot chew or swallow may require insertion of a feeding tube. Non-invasive ventilation at night can improve breathing during sleep, and some individuals also may require assisted ventilation during the day due to muscle weakness in the neck, throat, and chest.