
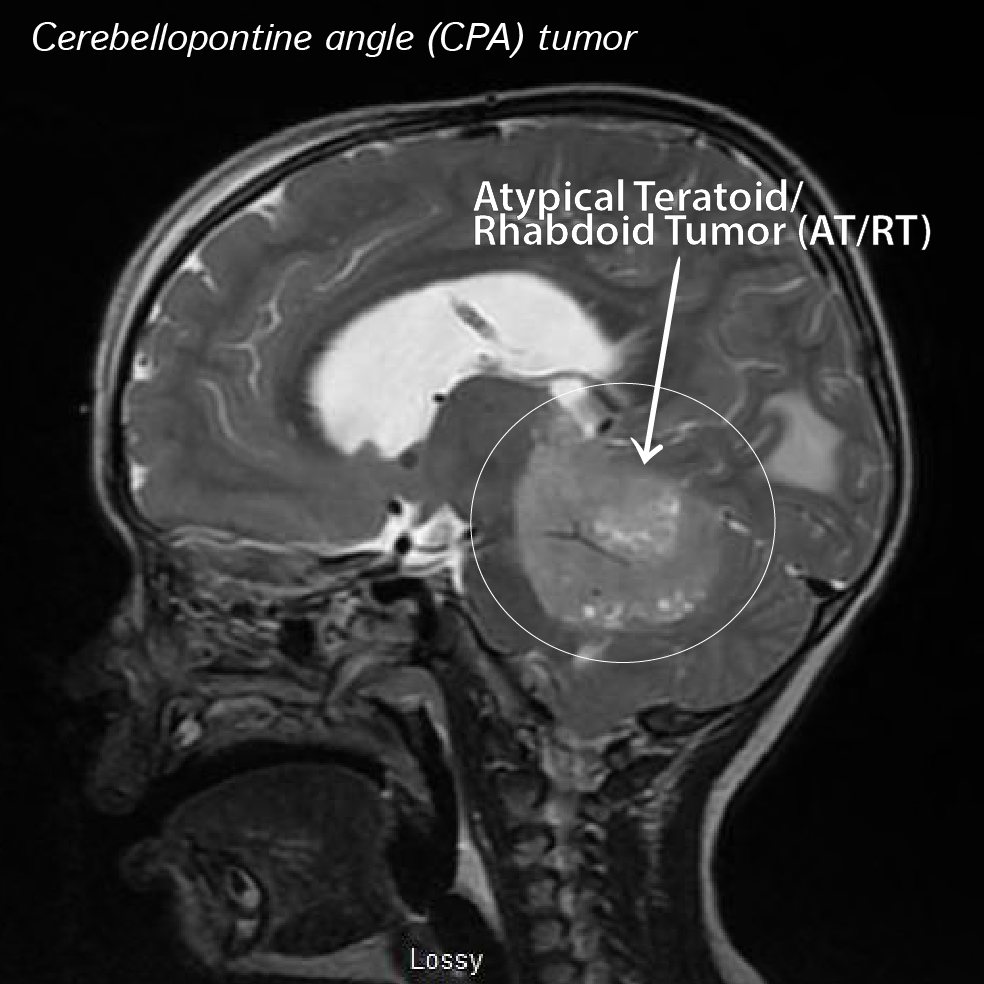
AT/RT is a primary central nervous system (CNS) tumor. This means it begins in the brain or spinal cord.
To get an accurate diagnosis, a piece of tumor tissue will be removed during surgery, if possible. A neuropathologist should then review the tumor tissue.
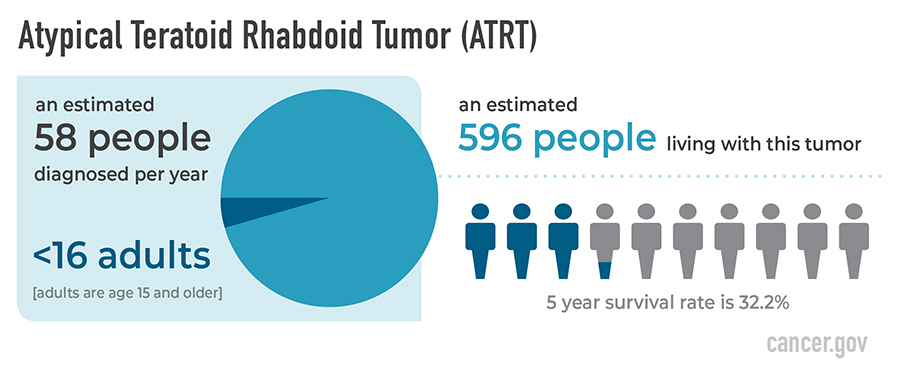
Central nervous system (CNS) atypical teratoid/rhabdoid tumor (AT/RT) is a very rare, fast-growing cancer that begins in the brain and spinal cord. It usually occurs in children aged 3 years and younger, although it can occur in older children and adults.
About half of these tumors form in the cerebellum or brain stem. The cerebellum is the part of the brain that controls movement, balance, and posture. The brain stem controls numerous functions breathing, heart rate, and the nerves and muscles used in seeing, hearing, walking, talking, and eating. AT/RT can also begin in other parts of the brain and spinal cord.
Certain genetic changes may increase the risk of AT/RT.
A risk factor is anything that increases the chance of getting a disease. Not every child with one or more of these risk factors will develop AT/RT. And it will develop in some children who don’t have a known risk factor.
AT/RT may be linked to changes in the tumor suppressor genes SMARCB1 or SMARCA4. Tumor suppressor genes make a protein that helps control how and when cells grow. Changes in the DNA of tumor suppressor genes like SMARCB1 or SMARCA4 may lead to cancer.
The changes in the SMARCB1 or SMARCA4 genes may be inherited (passed on from parents to offspring). When this gene change is inherited, tumors may form in two parts of the body at the same time (for example, in the brain and the kidney). For children with AT/RT, genetic counseling (a discussion with a trained professional about inherited diseases and a possible need for gene testing) may be recommended.
Most AT/RTs are caused by changes in a gene known as SMARCB1 (also called INI1), and less frequently by mutations in a gene called SMARCA4. SMARCB1 normally signals proteins to stop tumor growth. But, in AT/RTs, SMARCB1 doesn’t function properly and tumor growth is uncontrolled. In addition to occurring in the tumor’s DNA, SMARCB1 and SMARCA4 can also be found in a person’s own DNA. There are three groups of AT/RTs based on their genetic alterations: AT/RT-TYR, AT/RT-SHH, and AT/RT-MYC. Each group tends to develop in a different location of the CNS and is more common in different age groups. AT/RT-MYC is the most frequent group in adults.
Talk with your child’s doctor if you think your child may be at risk.
Cancer is a genetic disease—that is, it is caused by certain changes to genes that control the way our cells function. Genes may be mutated (changed) in many types of cancer, which can increase the growth and spread of cancer cells.
The Grades of the tumors:
Primary CNS tumors are graded based on the tumor location, tumor type, extent of tumor spread, genetic findings, the patient’s age, and tumor remaining after surgery, if surgery is possible.
AT/RTs are all classified as grade 4 (also written as grade IV) tumors. This means they are malignant (cancerous) and fast-growing.
The symptoms of AT/RT aren’t the same in each patient.
Symptoms depend on 2 major factors:
- the child’s age
- where the tumor has formed
Because AT/RT is fast growing, symptoms may develop quickly and get worse over a period of days or weeks. It’s important to check with your child’s doctor if your child has:
- a morning headache or headache that goes away after vomiting
- nausea and vomiting
- unusual sleepiness or change in activity level
- loss of balance, lack of coordination, or trouble walking
- an increase in head size (in infants)
- pain, tingling, numbness, or paralysis in the face
These symptoms may be caused by problems other than AT/RT. The only way to know is to see your child’s doctor.
The prognosis of this disease:
The likely outcome of the disease or chance of recovery is called prognosis. Prognosis is based on the tumor grade, location, tumor type, extent of tumor spread, genetic findings, patient’s age, and tumor remaining after surgery (if surgery is possible).
Many factors can affect prognosis, including the tumor grade and molecular type, the person’s age and health when diagnosed, and how they respond to treatment. If you want to understand your prognosis, talk to your doctor.
The Treatment of Teratoid/Rhabdoid Tumors (AT/RT):
The first treatment for an AT/RT is surgery, if possible. The goal of surgery is to obtain tissue to determine the tumor type and remove as much tumor as possible without causing more symptoms.
People with AT/RTs usually receive further treatments after surgery, which may include radiation, chemotherapy, or clinical trials. Clinical trials test new chemotherapy, targeted therapy, or immunotherapy drugs. Treatments are decided by the patient’s health care team based on the patient’s age, tumor remaining after surgery, tumor type, and tumor location.